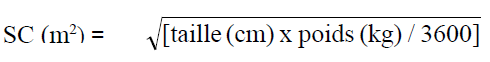- Imprimer
- Theme par defaut
- Theme doc avenue
- Theme dexther web
KALETRA 200 mg/50 mg, comprimé pelliculé, boîte de 1 flacon de 120

|
Comprimé pelliculé rouge gravé du [logo ABBOTT] et de « AL ». |
|
Chaque comprimé pelliculé de Kaletra contient 200 mg de lopinavir associé à 50 mg de ritonavir qui agit en potentialisant la pharmacocinétique du lopinavir. Pour la liste complète des excipients, voir rubrique Liste des excipients. |
|
Les comprimés contiennent : Pelliculage :
|
|
Kaletra est indiqué en association avec d'autres médicaments antirétroviraux pour le traitement des adultes, des adolescents et des enfants âgés de plus de deux ans infectés par le virus de l'immunodéficience humaine (VIH-1). Chez les patients infectés par le VIH-1 et déjà traités par des inhibiteurs de protéase, le recours au Kaletra devrait être basé sur les résultats des tests individuels de résistance virale et sur l'historique du traitement des patients (voir rubriques Mises en garde et précautions d'emploi et Propriétés pharmacodynamiques). |
|
Kaletra doit être prescrit par
des médecins expérimentés dans la prise en charge de l'infection par le VIH. Kaletra comprimé doit être
avalé en entier sans être ni mâché, ni coupé, ni broyé. Posologie Adultes et
adolescents Enfants
(âgés de 2 ans et plus) *La surface
corporelle peut être calculée grâce à l'équation suivante :
Enfants de
moins de 2 ans Traitement co-administré : Efavirenz ou névirapine
* Les
comprimés Kaletra ne doivent pas être mâchés, ni
coupés, ni broyés. Insuffisance
hépatique Insuffisance
rénale · Aucun ajustement posologique de lopinavir/ritonavir n'est nécessaire pendant la grossesse et le postpartum. · L'administration en une prise par jour de lopinavir/ritonavir n'est pas recommandée chez la femme enceinte en raison du manque de données cliniques et pharmacocinétiques. Mode
d'administration Les
comprimés Kaletra sont administrés par voie orale et
doivent être avalés en entier sans être ni mâchés, ni coupés, ni broyés. Les
comprimés Kaletra peuvent être pris au cours ou en
dehors d'un repas. | |||||||||||||
|
Hypersensibilité aux substances actives ou à l'un des excipients mentionnés à la rubrique Liste des excipients. Insuffisance hépatique sévère. Kaletra contient du lopinavir et du ritonavir qui sont tous deux des inhibiteurs de l'isoforme CYP3A du cytochrome P450. Kaletra ne doit pas être associé aux médicaments dont le métabolisme est fortement dépendant de l'isoforme CYP3A et pour lesquels des concentrations plasmatiques élevées sont associées à des effets indésirables graves ou engageant le pronostic vital. Ces médicaments sont notamment :
| Contre-indiqué dans les cas suivants :
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Patients
présentant des pathologies associées Insuffisance
hépatique Les patients
ayant des troubles pré-existants de la fonction hépatique, y compris une
hépatite chronique active ont, au cours d'un traitement par association
d'antirétroviraux, une fréquence plus élevée d'anomalies de la fonction hépatique
et doivent faire l'objet d'une surveillance appropriée. Chez ces patients, en
cas d'aggravation confirmée de l'atteinte hépatique, l'interruption ou l'arrêt
du traitement devra être envisagé. Une
augmentation des transaminases associée ou non à une élévation de la bilirubine
a été rapportée chez des patients mono-infectés par le VIH-1 ainsi que chez des
sujets traités en prophylaxie post-exposition. Ces anomalies ont été observées
au plus tôt dans un délai de 7 jours après l'instauration de lopinavir/ritonavir
en association avec d'autres antirétroviraux. Dans certains cas, le
dysfonctionnement hépatique était grave. Des examens de
laboratoire appropriés doivent être réalisés avant l'initiation du traitement
par lopinavir/ritonavir et une surveillance étroite doit être effectuée pendant
le traitement. Insuffisance
rénale Hémophilie Pancréatite La pancréatite
doit être envisagée si des symptômes cliniques (nausées, vomissements, douleurs
abdominales) ou des anomalies biologiques (telles qu'une augmentation de
l'amylase ou de la lipase sérique) évocateurs d'une pancréatite surviennent.
Les patients qui manifestent ces signes ou symptômes doivent être surveillés et
le traitement par Kaletra doit être arrêté si le diagnostic de pancréatite est
posé (voir rubrique Effets indésirables). Syndrome
Inflammatoire de Reconstitution Immunitaire Des maladies
auto-immunes (telles que la maladie de Basedow et l'hépatite auto-immune) ont
aussi été rapportées dans le cadre de la reconstitution immunitaire ;
cependant, le délai d'apparition est plus variable et les manifestations
cliniques peuvent survenir de nombreux mois après l'instauration du traitement. Ostéonécrose Allongement
de l'intervalle PR Poids
corporel et paramètres métaboliques Interactions
médicamenteuses Les
inhibiteurs puissants du CYP3A4 tels que les inhibiteurs de protéase peuvent
augmenter l'exposition à la bédaquiline, ce qui pourrait potentiellement
augmenter le risque d'effets indésirables liés à la bédaquiline. Par
conséquent, l'association de la bédaquiline et du lopinavir/ritonavir doit être
évitée. Cependant, si le bénéfice l'emporte sur le risque, la co-administration
de bédaquiline et de lopinavir/ritonavir doit être réalisée avec prudence. Une
surveillance plus fréquente de l'électrocardiogramme et une surveillance des
transaminases sont recommandées (voir rubrique Interactions avec d'autres
médicaments et autres formes d'interactions et se référer au RCP de la
bédaquiline). Une
administration concomitante de délamanide avec un inhibiteur puissant du CYP3A
(tel que lopinavir/ritonavir) est susceptible d'augmenter l'exposition au
métabolite du délamanide, ce qui a été associé à un allongement de l'intervalle
QTc. Par conséquent, si une administration concomitante de délamanide avec du
lopinavir/ritonavir est considérée comme nécessaire, il est recommandé
d'effectuer une surveillance très fréquente par ECG pendant toute la période de
traitement par délamanide (voir rubrique Interactions avec d'autres
médicaments et autres formes d'interactions et se reporter au RCP du
délamanide). Des
interactions médicamenteuses mettant en jeu le pronostic vital et fatales ont
été rapportées chez des patients traités avec de la colchicine et des
inhibiteurs puissants du CYP3A comme le ritonavir. L'administration
concomitante avec la colchicine est contre-indiquée chez les patients
présentant une insuffisance rénale et/ou hépatique (voir rubriques Contre-indications
et Interactions avec d'autres médicaments et autres formes d'interactions). · le tadalafil, indiqué dans le traitement de l'hypertension artérielle pulmonaire, n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) ; · le riociguat n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) ; · le vorapaxar n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) ; · l'acide fusidique utilisé dans les infections ostéo-articulaires n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) ; · le salmétérol n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) ; · le rivaroxaban n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). L'association
de Kaletra avec l'atorvastatine n'est pas recommandée. Si l'utilisation de
l'atorvastatine est considérée comme strictement nécessaire, la plus faible
dose possible d'atorvastatine doit être administrée avec une surveillance
accrue des effets indésirables. La prudence est également recommandée et des
réductions posologiques doivent être envisagées si Kaletra est utilisé en même
temps que la rosuvastatine. Si un traitement par un inhibiteur de l'HMG CoA
réductase est indiqué, la pravastatine ou la fluvastatine sont recommandées
(voir rubrique Interactions avec d'autres médicaments et autres formes
d'interactions). Inhibiteurs
de la phosphodiestérase 5 (PDE5) Une
surveillance particulière doit être mise en ?uvre lors de la prescription de
Kaletra avec des médicaments connus pour induire un allongement de l'intervalle
QT comme la chlorphéniramine, la quinidine, l'érythromycine, la
clarithromycine. En effet, Kaletra peut augmenter les concentrations
plasmatiques des médicaments co-administrés, ce qui peut provoquer une
augmentation des effets indésirables cardiaques associés. Des évènements
cardiaques ont été rapportés avec Kaletra au cours des études précliniques ; à
ce jour, les effets cardiaques potentiels de Kaletra ne peuvent pas être exclus
(voir rubriques Effets indésirables et Données de sécurité
précliniques). La
co-administration de Kaletra et de la rifampicine n'est pas recommandée. La
rifampicine associée à Kaletra provoque des diminutions importantes des
concentrations plasmatiques de lopinavir qui peuvent diminuer significativement
l'efficacité du lopinavir. Une exposition suffisante au lopinavir/ritonavir
peut être obtenue avec une augmentation de la dose de Kaletra mais les risques
de toxicités hépatique et gastro-intestinale sont alors augmentés. Par
conséquent, la co-administration doit être évitée à moins qu'elle ne soit
strictement nécessaire (voir rubrique Interactions avec d'autres médicaments
et autres formes d'interactions). L'utilisation
concomitante de Kaletra et de fluticasone, ou d'autres glucocorticoïdes
métabolisés par le CYP3A4, comme le budésonide et la triamcinolone, n'est pas
recommandée, à moins que le bénéfice attendu pour le patient ne l'emporte sur
le risque d'effets systémiques de la corticothérapie, tels qu'un syndrome de
Cushing ou une inhibition de la fonction surrénalienne (voir rubrique Interactions
avec d'autres médicaments et autres formes d'interactions). Autres |
|
|
Kaletra contient du lopinavir et du ritonavir qui sont tous deux des inhibiteurs de l'isoforme CYP3A du cytochrome P450 in vitro. La co-administration de Kaletra et des médicaments principalement métabolisés par l'isoforme CYP3A peut provoquer une augmentation des concentrations plasmatiques des médicaments associés, ce qui est susceptible d'augmenter ou de prolonger leurs effets thérapeutiques et leurs effets indésirables. Kaletra n'inhibe pas les isoformes CYP2D6, CYP2C9, CYP2C19, CYP2E1, CYP2B6 ou CYP1A2 aux concentrations thérapeutiques (voir rubrique Contre-indications). Il a été observé in vivo que Kaletra induit son propre métabolisme et qu'il augmente la biotransformation de certains médicaments métabolisés par le système enzymatique du cytochrome P450 (dont les isoformes CYP2C9 et CYP2C19) et par glucuronoconjugaison. Cela peut se traduire par des baisses des concentrations plasmatiques et une diminution de l'efficacité des médicaments associés. Les médicaments spécifiquement contre-indiqués, en raison de l'importance attendue de leur interaction et de la possibilité de survenue d'effets indésirables graves, sont présentés dans la rubrique Contre-indications. Toutes les études d'interaction, en l'absence d'indication contraire, ont été réalisées avec Kaletra capsules molles qui produit une exposition au lopinavir d'environ 20 % inférieure à celle produite par les comprimés à 200/50 mg. Les interactions connues et potentielles avec certains antirétroviraux et des médicaments autres que des antirétroviraux sont décrites dans le tableau ci-dessous. Cette liste n'est pas exhaustive ou complète. Le RCP de chacun des produits doit être consulté. Tableau des interactions Les interactions entre Kaletra et les médicaments co-administrés sont décrites dans le tableau ci- dessous (« ↑ » signifie « augmentation », « ↓ » signifie « diminution », « ↔ » signifie « pas de changement », « 1x/j » signifie « une fois par jour », « 2x/j » signifie « deux fois par jour », « 3x/j » signifie « trois fois par jour »). Sauf mention particulière, les études décrites ci-dessous ont été réalisées avec la posologie recommandée de lopinavir/ritonavir (c'est-à-dire 400/100 mg deux fois par jour).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Grossesse Le lopinavir/ritonavir a été évalué
chez plus de 3 000 femmes pendant la grossesse, dont plus de 1 000 au cours du
premier trimestre. Dans le cadre
de la surveillance après commercialisation provenant du registre de suivi des
grossesses sous antirétroviraux, mis en place depuis janvier 1989, aucune augmentation
du risque de malformations congénitales suite à une exposition à Kaletra n'a été rapportée chez plus de1 000 femmes exposées
au cours du 1 er trimestre. La prévalence des malformations congénitales suite
à une exposition au lopinavir, quel que soit le
trimestre, est comparable à celle observée dans la population générale. Aucun
type de malformations congénitales évocateur d'une étiologie commune n'a été
observé. Les études chez l'animal ont montré une toxicité sur la reproduction (voirrubrique Données de sécurité préclinique). Sur
la base des données disponibles, le risque malformatif est peu probable dans
l'espèce humaine. Le lopinavir peut être utilisé pendant la grossesse si
nécessaire. Allaitement Fertilité |
|
Les effets de Kaletra sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés. Les patients doivent être informés que des nausées ont été rapportées lors de traitements par Kaletra (voir rubrique Effets indésirables). |
a. Résumé du profil de sécurité La sécurité
d'emploi de Kaletra a été évaluée au cours d'études
cliniques de phase II-IV chez plus de 2 600 patients, dont plus de 700 ont reçu
une posologie de 800/200 mg (6 capsules ou 4 comprimés) une fois par jour. Dans
certaines études, avec les inhibiteurs nucléosidiques
de la transcriptase inverse (INTI), Kaletra était
également associé à l'éfavirenz ou à la névirapine. La diarrhée,
les nausées et vomissements, l'hypertriglycéridémie et l'hypercholestérolémie
ont été les effets indésirables les plus fréquemment liés au traitement par Kaletra durant ces études. Le risque de diarrhée peut être
plus élevé avec la posologie de Kaletra en une prise
par jour. La diarrhée, les nausées et les vomissements peuvent survenir au
début du traitement tandis que l'hypertriglycéridémie et l'hypercholestérolémie
peuvent survenir plus tardivement. La survenue d'effets indésirables liés au
traitement a conduit à une sortie prématurée des études de phase II-IV pour 7 %
des patients. Il est
important de noter que des cas de pancréatites ont été rapportés chez les
patients traités par Kaletra, parmi lesquels certains
présentaient une hypertriglycéridémie. De plus, de rares augmentations
de l'intervalle PR ont été rapportées pendant le traitement par Kaletra (voir rubrique Mises en garde et précautions
d'emploi.). b. Tableau des effets indésirables Effets
indésirables suite aux études cliniques et à l'expérience après
commercialisation chez les patients adultes et enfants : Les évènements
suivants ont été identifiés comme effets indésirables. Le classement par
fréquence comprend tous les effets indésirables rapportés, d'intensité modérée
à sévère, quelle que soit l'évaluation individuelle de la cause. Les effets
indésirables sont répertoriés par classe de systèmes d'organes. Au sein de
chaque groupe de fréquence, les effets indésirables sont présentés suivant un
ordre décroissant de gravité : très fréquent (≥ 1/10), fréquent (≥
1/100, < 1/10), peu fréquent (≥ 1/1
000, < 1/100), rare (≥ 1/10 000, < 1/1 000) et fréquence
indéterminée (ne peut être estimée sur la base des données disponibles).
1 voir rubrique Mises
en garde et précautions d'emploi : pancréatites et lipides c. Description d'effets indésirables particuliers Le syndrome de
Cushing a été rapporté chez des patients recevant du ritonavir
et du propionate de fluticasone
administré par inhalation ou par voie intranasale ; cet effet indésirable
pourrait également survenir avec d'autres corticostéroïdes métabolisés par le
cytochrome P450 3A par exemple le budésonide (voir
rubriques Mises en garde et précautions d'emploi et Interactions avec
d'autres médicaments et autres formes d'interactions). Une
augmentation de la créatine phosphokinase (CPK), des
myalgies, des myosites et rarement des rhabdomyolyses ont été rapportées avec
les inhibiteurs de protéase, et particulièrement en association avec les
inhibiteurs nucléosidiques de la transcriptase
inverse. Paramètres
métaboliques Chez les
patients infectés par le VIH et présentant un déficit immunitaire sévère au
moment de l'instauration du traitement par une association d'antirétroviraux,
une réaction inflammatoire à des infections opportunistes asymptomatiques ou résiduelles
peut se produire. Des maladies auto-immunes (telles que la maladie de Basedow
et l'hépatite auto-immune) ont aussi été rapportées ; cependant, le délai
d'apparition est plus variable et les manifestations cliniques peuvent survenir
de nombreux mois après l'instauration du traitement (voir rubrique Mises en
garde et précautions d'emploi). Des cas
d'ostéonécrose ont été rapportés, particulièrement chez des patients avec des
facteurs de risque généralement connus, une infection avancée par le VIH ou une
exposition à long terme au traitement par association d'antirétroviraux. La
fréquence de cet effet indésirable est indéterminée (voir rubrique Mises en
garde et précautions d'emploi). d. Population pédiatrique Chez les
enfants de deux ans et plus, le profil de sécurité d'emploi est similaire à
celui observé chez les adultes (voir tableau à la section b). Déclaration
des effets indésirables suspectés |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
L'expérience chez l'Homme de surdosage aigu avec Kaletra est limitée. Les signes de toxicité observés chez les chiens comprennent une salivation, des vomissements, des diarrhées et/ou des selles anormales. Les signes de toxicité observés chez les souris, les rats, les chiens comprennent une diminution de l'activité, une ataxie, une émaciation, une déshydratation et des tremblements. Il n'existe pas d'antidote spécifique de Kaletra. Le traitement du surdosage est symptomatique et nécessite la surveillance des fonctions vitales et de l'état clinique du patient. L'élimination de la substance active non absorbée pourra être réalisée, le cas échéant, par un lavage gastrique ou en administrant des émétiques. L'administration de charbon activé peut également être utilisée pour aider l'élimination de la substance non absorbée. Kaletra étant fortement lié aux protéines plasmatiques, l'intérêt de la dialyse pour éliminer une quantité significative de substance active est improbable. |
Classe pharmacothérapeutique : antiviraux pour usage systémique,
antiviraux pour le traitement des infections par le VIH, associations, code ATC
: J05AR10 Mécanisme
d'action Effets sur
l'électrocardiogramme Un léger
allongement de l'intervalle PR a également été observé le 3ème jour chez les
sujets recevant du lopinavir/ritonavir
dans la même étude. Les modifications moyennes de l'intervalle PR par rapport
aux valeurs initiales se sont réparties entre 11,6 ms et 24,4 ms dans les 12
heures post-dose. L'intervalle
PR maximal a été de 286 ms et aucun sujet n'a présenté de bloc cardiaque du
2ème ou du 3ème degré (voir rubrique Mises en garde et précautions d'emploi). Activité
antivirale in vitro Résistance Analyse de
la résistance chez des patients naïfs aux ARV Analyse de
la résistance chez des patients pré-traités par un IP Analyse de la
sensibilité génotypique des souches virales de sensibilité phénotypique
diminuée au lopinavir, sélectionnées par les autres
inhibiteurs de protéase. L'activité antivirale du lopinavir
in vitro sur 112 isolats cliniques de patients en échec d'une
monothérapie ou d'un traitement avec une ou plusieurs antiprotéases
a été évaluée. Dans cet échantillon, les mutations suivantes du gène de la
protéase du VIH ont été associées à une réduction in vitro de la
sensibilité au lopinavir : L10F/I/R/V, K20M/R, L24I,
M46I/L, F53L, I54L/T/V, L63P, A71I/L/T/V, V82A/F/T, I84V et L90M. La médianede la CE 50 du lopinavir
pour ces isolats présentant de 0 à 3, de 4 à 5, de 6 à 7 et de 8 à 10 mutations
sur les positions des acides aminés citées précédemment, a été respectivement
de 0,8 ; 2,7 ; 13,5 et 44,0 fois supérieure à la CE50 pour le virus de type
sauvage. Les 16 souches virales ayant montré une modification supérieure à 20
fois de leur sensibilité comportaient toutes des mutations au niveau des
positions 10, 54, 63 et 82 et/ou 84. De plus, une médiane de 3 mutations a été
observée au niveau des acides aminés positionnés en 20, 24, 46, 53, 71 et 90.
En plus des mutations décrites ci-dessus, les mutations V32I et I47A ont été
observées dans les cas de rebond virologique avec une réduction de la
sensibilité au lopinavir chez des patients pré-traités par un inhibiteur de protéase recevant un
traitement par Kaletra et les mutations I47A et L76V
ont été observées sur des isolats de rebond virologique présentant une
réduction de la sensibilité au lopinavir chez des
patients recevant un traitement par Kaletra. Les
conclusions concernant la pertinence de chaque mutation ou des profils de
mutations sont sujettes à modification en fonction de données futures, et il
est recommandé de toujours consulter les règles d'interprétation en vigueur
pour analyser les résultats d'un test de résistance. Activité
antivirale de Kaletra chez les patients en échec d'un
traitement par un inhibiteur de protéase Résistance
croisée Résultats
cliniques Chez
l'adulte Patients naïfs
de tout traitement antirétroviral L'étude
M98-863 était une étude randomisée en double aveugle menée chez 653 patients
naïfs de traitement antirétroviral recevant Kaletra
(400/100 mg, deux fois par jour) ou nelfinavir (750
mg trois fois par jour) en association à la stavudine
et la lamivudine. La valeur initiale moyenne du taux
de cellules T CD4+ était de 259 cellules/mm3 (de 2 à 949 cellules/mm3) et la
valeur moyenne initiale de la charge virale était de 4,9 log10 copies/ml (de
2,6 à 6,8 log10 copies/ml). Tableau 1
*analyse en
intention de traiter : patients avec valeurs manquantes considérés en échec
virologique Cent treize
patients traités par nelfinavir et 74 patients
traités par lopinavir/ritonavir
avaient une charge virale supérieure à 400 copies/ml bien que sous traitement
de la semaine 24 à la semaine 96. Parmi ces patients, les isolats de 96
patients traités par nelfinavir et 51 patients
traités par lopinavir/ritonavir
ont pu être amplifiés pour test de résistance. Une résistance au nelfinavir, définie par la présence de la mutation D30N ou
L90M au niveau de la protéase, a été observée chez 41/96 (43 %) patients. Une
résistance au lopinavir, définie par la présence de
mutations sur le site primaire ou actif de la protéase (voir ci-dessus) a été
observée chez 0/51 (0 %) patients. L'absence de résistance au lopinavir a été confirmée par analyse phénotypique. L'étude
M05-730 était une étude randomisée, en ouvert, multicentrique évaluant le
traitement par Kaletra 800/200 mg une fois par jour
par rapport à Kaletra 400/100 mg deux fois par jour,
en association avec le ténofovir TDF (300 mg une fois
par jour, équivalent à 245 mg de ténofovir disoproxil) et l'emtricitabine
(200 mg une fois par jour), chez 664 patients naïfs de traitement
antirétroviral. Du fait de l'interaction pharmacocinétique entre Kaletra et le ténofovir (voir
rubrique Interactions avec d'autres médicaments et autres formes
d'interactions), les résultats de cette étude pourraient ne pas être
strictement extrapolables lorsque d'autres médicaments antirétroviraux sont
associés à Kaletra. Les patients ont été randomisés
(1:1) pour recevoir Kaletra 800/200 mg une fois par jour (333 patients) ou Kaletra 400/100 mg deux fois par jour (331 patients). Une
stratification supplémentaire a été réalisée dans chacun des groupes (comprimé versus
capsule molle, 1:1). Pendant les 8 premières semaines de l'étude, les
patients ont reçu soit les comprimés, soit les capsules molles puis tous les
patients ont reçu les comprimés en une ou deux fois par jour jusqu'à la fin de
l'étude. Selon le protocole, la non-infériorité de l'administration en une
prise par jour par rapport à celle en deux prises par jour était démontrée si
la limite inférieure de l'intervalle de confiance (IC à 95 %) pour la
différence de pourcentage des répondeurs (« une fois par jour » moins « deux fois
par jour ») excluait -12 % à la semaine 48. L'âge moyen des patients inclus
était de 39 ans (de 19 à 71 ans) ; 75 % étaient de type Caucasien et 78 % des
hommes. La valeur initiale moyenne du taux de cellules T CD4+ était de 216
cellules/mm3 (de 20 à 775 cellules/mm3) et celle de la charge virale de 5,0
log10 copies/ml (de 1,7 à 7,0 log10 copies/ml). Tableau 2
Au cours des
96 semaines de traitement, les résultats des tests génotypiques de résistance
ont été disponibles pour 25 patients du groupe « une fois par jour » et pour 26
patients du groupe « deux fois par jour » qui étaient en échec virologique.
Dans le groupe « une fois par jour », aucun patient n'a présenté de résistance
au lopinavir ; dans le groupe « deux fois par jour »,
1 patient qui avait une résistance primaire à l'inhibiteur de protéase à
l'initiation du traitement, a présenté une résistance additionnelle au lopinavir durant l'étude. Une réponse
virologique prolongée à Kaletra (associé à des
inhibiteurs nucléosidiques/nucléotidiques de la
transcriptase inverse) a également été observée au cours d'une étude de phase
II (M97-720) à 360 semaines de traitement. Cent patients ont été initialement
traités par Kaletra au cours de l'étude (dont 51
patients par 400/100 mg deux fois par jour et 49 patients soit par 200/100 mg
deux fois par jour soit par 400/200 mg deux fois par jour). Tous les patients
sont passés à la dose de Kaletra de 400/100 mg deux
fois par jour, en ouvert, entre la semaine 48 et la semaine 72. Trente-neuf
patients (39 %) ont arrêté l'étude, 16 d'entre eux (16 %) en raison d'effets
indésirables, dont un associé au décès du patient. Soixante-et-un patients ont
terminé l'étude (35 patients ont reçu la dose recommandée de 400/100 mg deux
fois par jour pendant toute la durée de l'étude). Tableau 3
Au cours des
360 semaines de traitement, l'analyse génotypique des isolats viraux conduite
avec succès chez 19 des 28 patients avec une charge virale confirmée supérieure
à 400 copies/ml n'a pas mis en évidence de mutation sur le site primaire ou
actif de la protéase (acides aminés en position 8, 30, 32, 46, 47, 48, 50, 82,
84 et 90) ou de résistance phénotypique à l'inhibiteur de la protéase. Patients pré-traités par des antirétroviraux L'étude
M06-802 était une étude randomisée, en ouvert comparant la sécurité d'emploi,
la tolérance et l'activité antirétrovirale de l'administration en une prise par
jour et deux prises par jour du lopinavir/ritonavir en comprimé chez 599 patients ayant une charge
virale détectable malgré un traitement antiviral en cours. Les patients
n'avaient jamais été pré-traités par le lopinavir/ritonavir. Ils ont été
randomisés (1:1) pour recevoir soit 800/200 mg de lopinavir/ritonavir en une prise par jour (n = 300) soit 400/100 mg
de lopinavir/ritonavir en
deux prises par jour (n = 299). Les patients avaient reçu également au moins
deux inhibiteurs nucléosidiques/nucléotidiques de la
transcriptase inverse sélectionnés par les investigateurs. La population
incluse dans l'étude était une population peu exposée aux inhibiteurs de la
protéase avec plus de la moitié des patients n'ayant jamais reçu d'inhibiteurs
de la protéase au préalable et environ 80 % des patients présentant une souche
virale avec moins de 3 mutations aux inhibiteurs de la protéase. L'âge moyen
des patients inclus était de 41 ans (de 21 à 73 ans) ; 51 % étaient de type
Caucasien et 66 % des hommes. À l'inclusion, la valeur moyenne du taux de
cellules T CD4+ était de 254 cellules/mm3 (de 4 à 952 cellules/mm3) et celle de
la charge virale était de 4,3 log10 copies/ml (de 1,7 à 6,6 log10 copies/ml).
Environ 85 % des patients avaient une charge virale < 100 000 copies/ml. Tableau 4
Au cours des
48 semaines de traitement, les résultats des tests génotypiques de résistance
ont été disponibles pour 75 patients du groupe « une fois par jour » et pour 75
patients du groupe « deux fois par jour » qui étaient en échec virologique.
Dans le groupe « une fois par jour », 6/75 (8 %) patients ont présenté de
nouvelles mutations primaires à l'inhibiteur de protéase (codons 30, 32, 48,
50, 82, 84, 90) comme 12/77 (16 %) patients dans le groupe « deux fois par jour
». Chez
l'enfant L'étude
M98-940 était une étude en ouvert réalisée chez 100 enfants avec la forme
liquide de Kaletra dont 44 % des patients étaient
naïfs et 56 % pré-traités. Tous les patients étaient
naïfs aux inhibiteurs non nucléosidiques de la
transcriptase inverse. Les patients ont été randomisés et répartis dans deux
groupes recevant soit 230 mg de lopinavir/57,5 mg de ritonavir par m2 de surface corporelle ou 300 mg de lopinavir/75 mg de ritonavir par
m2 de surface corporelle. Les patients naïfs ont également reçu des inhibiteurs
nucléosidiques de la transcriptase inverse. Les
patients pré-traités ont reçu de la névirapine et jusqu'à deux inhibiteurs nucléosidiques
de la transcriptase inverse. La sécurité, l'efficacité et le profil
pharmacocinétique des deux schémas posologiques ont été évalués après 3
semaines de traitement pour chaque patient. Par la suite, tous les patients ont
continué avec une posologie de 300/75 mg par m2 de surface corporelle. Les
patients présentaient un âge moyen de 5 ans (de 6 mois à 12 ans), 14 patients
avaient moins de 2 ans et 6 patients un an ou moins. La valeur moyenne du taux
initial de cellules T CD4+ était de 838 cellules/mm3 et la charge virale
plasmatique initiale avait une valeur moyenne de 4,7 log10 copies/ml. Tableau 5
L'étude KONCERT/PENTA
18 est une étude prospective, multicentrique, randomisée, en ouvert, qui a
évalué le profil pharmacocinétique, l'efficacité et la sécurité d'emploi de
l'administration en deux prises par jour versus une prise par jour de
comprimés à 100 mg/25 mg de lopinavir/ritonavir, les doses étant administrées en fonction du
poids, dans le cadre d'un traitement par association d'antirétroviraux chez des
enfants infectés par le VIH-1 et virologiquement
contrôlés (n = 173). Les enfants étaient éligibles s'ils étaient âgés de moins
de 18 ans, d'un poids ≥ 15 kg, sous traitement par association
d'antirétroviraux incluant le lopinavir/ritonavir, avec une charge virale < 50 copies/ml depuis
au moins 24 semaines et capables d'avaler des comprimés. À la semaine 48,
l'efficacité et la sécurité d'emploi de l'administration chez l'enfant de
comprimés à 100 mg/25 mg de lopinavir/ritonavir en deux prises par jour (n = 87) étaient
similaires aux résultats obtenus dans les études déjà réalisées chez l'adulte
et chez l'enfant avec une administration de lopinavir/ritonavir en deux prises par jour. Le pourcentage de
patients avec un rebond virologique confirmé > 50 copies/ml durant les 48
semaines de suivi était supérieur chez les enfants recevant lopinavir/ritonavir en une prise par jour (12%) que chez les patients
recevant la dose en deux prises par jour (8%, p = 0, 19), principalement
en raison d'une plus faible observance dans le groupe en une prise par jour.
Les différences de paramètres pharmacocinétiques entre ces deux schémas
d'administration renforcent les données d'efficacité en faveur d'une
administration en deux prises par jour (voir rubrique Propriétés
pharmacocinétiques). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Infectiologie - Parasitologie Antiviraux systémiques Inhibiteurs de protéase Association d'inhibiteurs de protéase Lopinavir + Ritonavir |
| J - Anti-infectieux généraux à usage systémique J05 - Antiviraux à usage systémique J05A - Antiviraux à action directe J05AR - Antiviraux pour le traitement des infections HIV en association J05AR10 - Lopinavir et ritonavir |
|
Les
propriétés pharmacocinétiques du lopinavir associé au ritonavir ont été
évaluées chez des adultes volontaires sains et chez des patients
infectés par le VIH ; aucune différence significative n'a été observée
entre les deux groupes. Le lopinavir est essentiellement métabolisé par
l'isoforme CYP3A. Le ritonavir inhibe le métabolisme du lopinavir, et
augmente donc les concentrations plasmatiques du lopinavir. Au cours
des essais cliniques, l'administration de Kaletra à la posologie de
400/100 mg deux fois par jour, chez les patients infectés par le VIH,
permet d'obtenir des concentrations moyennes de lopinavir à l'état
d'équilibre de 15 à 20 fois supérieures à celles du ritonavir. Les taux
plasmatiques de ritonavir représentent moins de 7 % de ceux obtenus
après une dose de ritonavir de 600 mg deux fois/jour. La CE50 antivirale in vitro du lopinavir est environ 10 fois inférieure à celle du ritonavir. Absorption Effets de l'alimentation sur l'absorption orale Distribution Biotransformation Élimination Administration en une prise par jour : les données pharmacocinétiques de Kaletra en une prise par jour ont été évaluées chez des patients infectés par le VIH naïfs de traitement antirétroviral. Kaletra 800/200 mg a été administré en association avec 200 mg d'emtricitabine et 300 mg de ténofovir DF, en une prise par jour pour les trois antirétroviraux. L'administration répétée de Kaletra 800/200 mg une fois par jour, pendant deux semaines, sans contrainte d'alimentation chez 16 patients, a conduit à un pic de concentration plasmatique (Cmax) moyenne du lopinavir de 14,8 + 3,5 ?g/ml, survenant 6 heures environ après la prise. La concentration résiduelle (Crés) moyenne à l'équilibre avant la prise du matin était de 5,5 + 5,4 ?g/ml. L'ASC sur 24h du lopinavir était de 206,5 + 89,7 ?g?h/ml. En comparaison avec l'administration en deux prises par jour, l'administration en une prise par jour est associée à une diminution d'environ 50 % des Cmin et Crés. Populations particulières Enfants Sexe, race et âge Grossesse et postpartum Dans une autre étude de pharmacocinétique en ouvert, 19 femmes enceintes infectées par le VIH ont reçu pendant la grossesse 400/100 mg de lopinavir/ritonavir deux fois par jour dans le cadre d'un traitement par association d'antirétroviraux débuté avant la conception. Des échantillons de sang ont été prélevés avant administration des doses (pré-doses) et à plusieurs reprises sur une période de 12 heures au trimestre 2 et au trimestre 3, à la naissance, et 4 à 6 semaines après l'accouchement (chez les femmes qui ont continué le traitement après l'accouchement) pour une analyse pharmacocinétique des concentrations plasmatiques des fractions totale et libre de lopinavir. Les données pharmacocinétiques chez les femmes enceintes infectées par le VIH-1 ayant reçu des comprimés à 400/100 mg de lopinavir/ritonavir deux fois par jour sont présentées dans le tableau 6 (voir rubrique Posologie et mode d'administration). Tableau 6
Insuffisance rénale Insuffisance hépatique
| |||||||||||||||||||||||||
|
Les études de toxicité à doses répétées chez les rongeurs et les chiens ont permis d'identifier les organes cibles principaux tels que le foie, les reins, la thyroïde, la rate et les hématies circulantes. Des modifications hépatiques à type d'hypertrophie cellulaire avec un phénomène de dégénérescence focale ont été observées. Pendant la période d'exposition, ces modifications ont été comparables ou inférieures à celles observées durant la période d'exposition clinique chez l'Homme, les doses chez l'animal étaient de plus de 6 fois celles de la dose thérapeutique recommandée. Une dégénérescence tubulaire rénale légère a été observée uniquement chez la souris pour une exposition égale au moins à deux fois la dose recommandée chez l'Homme ; les reins n'ont pas été endommagés chez les rats et les chiens. La réduction de la thyroxine sérique a entraîné une augmentation de la libération de la TSH aboutissant à une hypertrophie de la cellule folliculaire de la glande thyroïde des rats. Ces modifications ont été réversibles lors de l'interruption de la substance active et n'ont pas été observées chez la souris ni chez le chien. Un test de Coombs négatif avec anisocytose et poïkilocytose a été observé chez le rat mais pas chez la souris ni chez le chien. Une splénomégalie avec histiocytose est apparue chez le rat, mais pas chez les autres espèces animales. Le cholestérol sérique a été augmenté chez les rongeurs mais pas chez les chiens, alors que les triglycérides ont été augmentés seulement chez la souris. Au cours d'études in vitro, les canaux potassiques cardiaques humains clonés (HERG) ont été inhibés de 30 % aux concentrations testées les plus élevées de lopinavir/ritonavir, celles-ci correspondent à une exposition de 7 fois les pics de concentrations plasmatiques des fractions totales de lopinavir et de 15 fois les pics de concentrations plasmatiques de la fraction libre de lopinavir atteints chez l'Homme à la dose thérapeutique maximale recommandée. En revanche, des concentrations similaires de lopinavir/ritonavir n'ont démontré aucun retard de repolarisation des fibres cardiaques de Purkinje de chien. Des concentrations plus basses de lopinavir/ritonavir n'ont pas entraîné de blocage significatif du courant potassique (HERG). Les études de distribution tissulaire conduites chez le rat ne suggèrent pas de rétention significative de la substance active au niveau cardiaque, l'ASC cardiaque à 72 heures représentait approximativement 50 % de l'ASC mesurée au niveau plasmatique. Par conséquent, il est raisonnable de s'attendre à ce que les concentrations cardiaques de lopinavir ne soient pas significativement supérieures aux concentrations plasmatiques. Chez le chien, des ondes U proéminentes ont été observées sur l'électrocardiogramme associées à un intervalle PR prolongé et à une bradycardie. Il a été suggéré que ces effets étaient attribués à un désordre électrolytique. La pertinence clinique de ces données pré-cliniques n'est pas connue, toutefois, les effets cardiaques potentiels du médicament chez l'Homme ne peuvent être exclus (voir rubriques Mises en garde et précautions d'emploi et Effets indésirables). Chez le rat, une embryo-f?totoxicité (avortement, diminution de la viabilité f?tale, diminution du poids du f?tus, augmentation des variations du squelette) et une toxicité du développement postnatal (diminution de la survie des ratons) ont été observées aux doses materno-toxiques. L'exposition systémique de lopinavir/ritonavir aux dosages materno-toxiques et toxiques pour le développement postnatal a été inférieure à l'exposition thérapeutique destinée à l'Homme. Des études de carcinogénèse à long terme de l'association lopinavir/ritonavir menées chez la souris ont révélé une induction mitogénique, non génotoxique, de tumeurs du foie généralement considérée comme ayant peu de pertinence sur le risque chez l'Homme. Des études de carcinogénèse menées chez le rat n'ont pas révélé de potentiel tumoral. L'association lopinavir/ritonavir ne s'est révélée ni mutagène ni clastogène sur une batterie de tests réalisés in vitro et in vivo comportant notamment le test d'Ames, le test sur lymphome de souris, le test du micronucléus sur la souris et le test d'aberrations chromosomiques sur cultures de lymphocytes humains. |
Sans objet. |
|
Durée de conservation : Conditionnements en flacon : 4 ans. Précautions particulières de conservation : Pas de précautions particulières de conservation.
|
| Flacon de polyéthylène haute densité (PEHD) muni d'un bouchon en propylène. Chaque flacon contient 120 comprimés. 1 flacon de 120 comprimés pelliculés. |
| Pas d'exigences particulières.
|
| Liste I. Prescription initiale hospitalière annuelle. |
| SURVEILLANCE du traitement : - biologique : cholestérolémie, triglycéridémie, glycémie, enzymes hépatiques. |
Bien qu'il ait été démontré que l'efficacité virologique d'un traitement antirétroviral réduise sensiblement le risque de transmission du VIH par voie sexuelle, un risque de transmission ne peut être exclu. Des précautions doivent être prises conformément aux recommandations nationales afin de prévenir toute transmission. PREVENIR LE MEDECIN en cas de : - étourdissement, évanouissement ou sensation de battements cardiaques anormaux. PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (nausées).
|
| Code CIP7 | 3760993 |
| Code CIP13 | 3400937609937 (Code 13 référent) |
| Code UCD7 | 9286995 |
| Code UCD13 | 3400892869957 |
| Code CIS | 67969992 |
| Médicament T2A | Non |
| Laboratoire titulaire AMM | ABBVIE DEUTSCH GMBH&CO |
| Laboratoire exploitant | ABBVIE 10, rue d'Arcueil 94528 RUNGIS CEDEX Tel : 01 45 60 13 00 Fax : 01 45 60 13 01 Site Web : www.abbvie.fr |
| Prix de vente TTC | 304.64 € (Prix hors honoraire de dispensation - pharmacie) |
| Taux de TVA | 2.1 % |
| Base de remboursement SS | 304.64 € (Prix hors honoraire de dispensation - pharmacie) |
| Taux SS | 100 % |
| Code Acte pharmacie | PH1 |
| Médicament d'exception | Non |
| Agrément collectivités | Oui |
| Liste | Liste I |
| Statut | Médicament - AMM Européenne |
| Date AMM | 27/06/2006 |
| Rectificatif d'AMM | 23/04/2021 |
| Numéros AMM | KALETRA 200 mg/50 mg, comprimé pelliculé, boîte de 1 flacon de 120 : EU/1/01/172/004 |
| Marque | KALETRA |
| Gamme | Sans gamme |